This is the second piece in a two-part conversation with the founders of Veda Scientific, CEO Leo Welder and CSO Aldwin M. Anterola, PhD. To read part one, click here.
In part one, we chatted about their backgrounds, their approach to cannabis testing, their role in the greater industry and how they came into the cannabis industry.
In part two, we’re going down a few cannabis chemistry rabbit holes and realizing that what we don’t know is a lot more than what we do know. Join us as we delve into the world of volatile compounds, winemaking, the tastes and smells of cannabis, chicken adobo and much more.
Aaron: Alright so you mentioned the GCxGC/MS and your more advanced terpene analysis. How do you envision that instrument and that data helping your customers and/or the industry?
Leo: Some of the things that we envision will help is a better understanding of what compounds and what ratios will lead to desirable outcomes, things like better effects, aroma and flavor. By better understanding these things it’ll help the industry create better products.
I have a personal connection to this. My wife has some insomnia and she’s always had to take various forms of OTC pharmaceuticals to help with sleep. She tried using a 1:1 vape pen and it was a miracle worker for her for several months. The local dispensary had a sale on it, and she bought some extra. Unfortunately, even though she used it the same way as before, she got very serious anxiety, which obviously didn’t help her sleep. Every time she used the vapes from this same batch, she felt the same extreme anxiety. Sadly, she now had a lot of this product that she couldn’t use because it kept her awake rather than helping her sleep, so she went back to trying other OTC solutions. That’s a problem for both consumers and the industry at large. If people find something that works and provides a desired effect, they need to be able to rely on that consistency every time they purchase the product, leading to similar outcomes and not exaggerating the problem. That’s why I think consistency is so important. We’re taking two steps forward and one back when we have inconsistent products. How do we really grow and expand the availability of cannabis if we lose trust from our consumer base? What a lab can do and what we can do is provide data to cultivators and manufacturers to create that consistency and ultimately allow the market to expand into other demographics that are currently wary and less tolerant of that variance.

On a similar note, we have been having a lot of discussions with the CESC [Clinical Endocannabinoid System Consortium] down in San Diego. They are an advanced cannabis research group that we have been working with for over a year. We’ve started looking at the idea of varietals. To be more specific, because I’m not a wine connoisseur, varietals are the pinot noirs, the cabernets and sauvignon blancs of the industry. In the cannabis industry, consumers have indica and sativa, though we still argue over what that concept really means, if anything. But for the sake of argument, let’s say we have this dichotomy to use as a foundational decision tool for consumers- call it the red and white wine of the cannabis industry. How inaccessible would wine be if we just had red or white? Imagine if you went to a dinner party, really liked the wine you were drinking, and the host could only tell you that it was a red wine. You can’t go to a wine store and expect to find something similar to that wine if the only information you have is “red.” At a minimum, you need a category. So that’s what varietals are, the categories. The data that we can produce could help people in the industry who identify and establish the varietals based on their expertise as connoisseurs and product experts to find what those differences are chemically. Similarly, we’re also looking at appellation designations in California. So, we want to help provide tools for farmers to identify unique characteristics in their flower that would give them ability to claim and prove appellation designation.
Aldwin: The GCxGC/MS allows us to find more things besides the typical terpene profile with 20 or 40 terpenes. It allows us to go beyond those terpenes. The issue sometimes is that with a typical one-dimensional GC method, sure you could probably separate and find more terpenes, but the one dimension is not enough to separate everything that coelutes. And it’s not just terpenes. Some terpenes coelute with one another and that’s why people can see this inconsistency. Especially if you use a detector like an FID, we can see the compound limonene on the chromatogram, but there’s another terpene in there that is unknown that coelutes with limonene. So, this instrument is helping us get past the coeluting issue and solve it so that we know what peaks represent what terpenes.
The other bonus with our GCxGC/MS is that the coeluting compounds that were masked behind other terpenes are now revealed. There is a second dimension in the chromatogram where we can now detect some compounds in cannabis that would be hiding behind these large peaks if it were just a one-dimensional GC. Besides terpenes, we’ve found esters, alkanes, fatty acids, ketones, alcohols and aldehydes, as well as thiols. The terpenes are so plentiful in cannabis that these other compounds present at lower levels cannot be seen with just one-dimensional GC. There are just so many compounds in cannabis that the ones in small amounts are often masked. My analogy to highlight the importance of these minor compounds is like a dish; I am from the Philippines and I like chicken adobo. My father does it differently from my mom and someone else will do it differently in a different region. The base of the sauce is vinegar and soy sauce, but some people will do it differently and maybe add some bay leaf, garlic, pepper, or a touch of another spice. It’s still chicken adobo, but it tastes differently. Just like in cannabis, where yes, you have the same amount of THC in two different plants, but it’s still giving you a different experience. Some people say it’s because of terpenes, which is true in a lot of cases, but there are a lot of other volatile compounds that would explain better why certain dishes taste different.
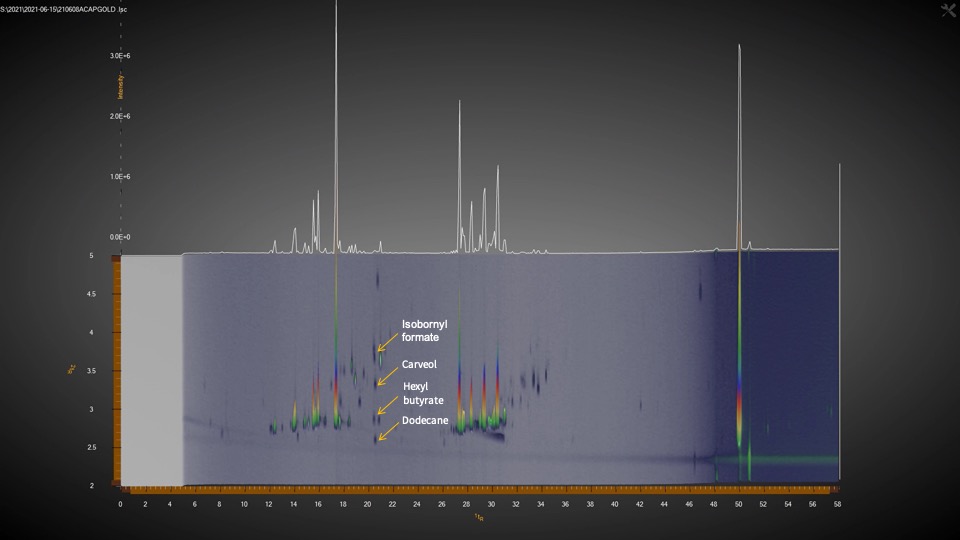
Leo: There’s been some recent developments too here that show it’s very significant. It’s like the difference between bland and spicy. And it could be the thiol. We identified a thiol in cannabis at the same time as other scientists reported an article that just came out on this subject.
Aldwin: Thiols are sulfur containing compounds that produce very powerful odors, giving cannabis the skunky smell. Skunks also produce thiols. It is very potent; you only need a little bit. It turns out that yes, that paper described thiols and we also saw them in our GCxGC/MS. These are the kinds of things that the GCxGC can show you. Those very tiny amounts of compounds that can have a very powerful impact. That’s one that we know for sure is important because it’s not just us that’s finding out that GCxGC can detect this.
Not everything is about THC or the high amount of the compounds in the flower. This paper and our concurrent findings indicated that the skunkier smelling strains contained very small amounts of thiols and you can recognize their presence quite readily. It’s not a terpene, but it’s producing a distinct flavor and a powerful smell.
Aaron: Okay, so why is this useful? Why is it so important?
Leo: I would say two things in particular that we know of that are issues currently, both related to scents. We mentioned this earlier. We do know that farmers with breeding programs are trying to target particularly popular or attractive scent profiles, whether it be a gas or fruity aroma. Right now, when they get the flower tested and review the terpene profile, it isn’t enough information to help them identify what makes them chemically distinct. We hear time and again that farmers will say their terpene profile is not helpful in identifying specific scents and characteristics. They are looking for a fingerprint. They want to be able to identify a group of plants that have a similar smell and they want a fingerprint of that plant to test for. Otherwise, you have to sniff every plant and smell the ones that are most characteristic of what they’re targeting. For larger operations, walking through and smelling thousands of plants isn’t feasible.
Once we can identify that fingerprint, and we know which compounds in which ratios are creating the targeted aroma, we can run tests to help them find the best plants for breeding purposes. It’s about reproducibility and scalability.
Another value is helping people who are trying to categorize oils and strains into particular odor categories, similar to the varietals concept we’ve been talking about. Currently, we know that when manufacturers send multiple samples of oils with the same or similar scent to be tested, the results are coming back with significantly different terpene profiles. There is not enough data for them to chemically categorize products. It’s not that their categories are wrong, it’s just that the data is not available to help them find those boundaries.
Those are two issues that we know from conversations with customers that this particular piece of equipment can address.
Aldwin: Let’s start from what we find, meaning if you are using the GCxGC/MS, we are finding more terpenes that nobody else would be looking at. We have data that shows, for example, that certain standards are accounting for 60% or so of total terpene content. So a large percent is accounted for, but there is still quite a bit missing. For some strains there are terpenes that are not in common reference standards. Being able to know that and identify the reason why we have different terpenes in here unaccounted for is big. There are other things there beyond the standard terpenes.

What excites me sometimes is that I see some terpenes that are known to have some properties, either medical or antibacterial, etc. If you find that terpene looking beyond the list, you’ll find terpenes that are found in things like hardwood or perfumes, things that we don’t necessarily associate with the common cannabis terpenes. If you’re just looking for the limited number of terpenes, you are missing some things that you might discover or some things that might help explain results.
Leo: It’s also absolutely necessary for the medical side of things. Because of the federal limitations, cannabis hasn’t been researched nearly enough. We’re missing a lot of data on all of the active compounds in cannabis. We are finally starting to move into an era where that will soon be addressed. In order for certain medical studies to be successful, we need to have data showing what compounds are in what plants.
Drs. John Abrams and Jean Talleyrand of the CESC launched the Dosing Project in 2016. They have been studying the impact of cannabis flower for indications such as pain mitigation and sleep improvement, and now more recently mood, and appetite modulation. They categorize the THC & CBD content as well as flower aroma into 3 cannabinoid and 3 odor profiles. They are able to acquire quite a bit of data about how odor correlates with the outcomes. Because they were initially limited in terms of underlying natural product content data, they contacted us when they found out we acquired this equipment in 2020, and have stated that they are certain the data we will now be producing will take their research to the next level of understanding.
 Aldwin: For quality control you are looking at specific things that would reflect properties in cannabis. There should be a 1:1 correspondence between properties observed and what we are measuring. The current assumption is that the terpenes we are looking at will tell us everything about how people would like it, with regards to flavor and smell preference. But we know for a fact that the limited terpenes most labs are measuring do not encapsulate everything. So, it is important for QC purposes to know for this particular strain or product, which everyone liked, what is it in there that makes everybody like it? If you just look at the typical terpene profile, you’ll find something close, but not exact. The GCxGC/MS shows us that maybe there’s something else that gives it a preferred property or a particular smell that we can explain and track. In one batch of flower, the consumer experiences it a certain way, and for another batch people experience it another way. We’d like to be able to understand what those differences are batch to batch so we can replicate the experience and figure out what’s in it that people like. That’s what I mean by consistency and quality control; the more you can measure, the more you can see.
Aldwin: For quality control you are looking at specific things that would reflect properties in cannabis. There should be a 1:1 correspondence between properties observed and what we are measuring. The current assumption is that the terpenes we are looking at will tell us everything about how people would like it, with regards to flavor and smell preference. But we know for a fact that the limited terpenes most labs are measuring do not encapsulate everything. So, it is important for QC purposes to know for this particular strain or product, which everyone liked, what is it in there that makes everybody like it? If you just look at the typical terpene profile, you’ll find something close, but not exact. The GCxGC/MS shows us that maybe there’s something else that gives it a preferred property or a particular smell that we can explain and track. In one batch of flower, the consumer experiences it a certain way, and for another batch people experience it another way. We’d like to be able to understand what those differences are batch to batch so we can replicate the experience and figure out what’s in it that people like. That’s what I mean by consistency and quality control; the more you can measure, the more you can see.
Aldwin: Speaking to authenticity as well, in a breeding example, some growers will have this strain that they grew, or at least this is what they claim it to be, but what are the components that make those strains unique? The more analytes you can detect, the more you can authenticate the plant. Is this really OG Kush? Is this the same OG Kush that I’ve had before? Using the GCxGC/MS and comparing analytes, we can find authenticity in strains by finding all of the metabolites and analytes and comparing two strains. Of course, there is also adulteration- Some people will claim they have one strain that smells like blueberries, but we find a compound in it that comes from outside of cannabis, such as added terpenes. Proving that your cannabis is actually pure cannabis or proving that something has added terpenes is possible because we can see things in there that don’t come from cannabis. The GCxGC/MS can be used as a tool for proving authenticity or proving adulteration as well. If you want to trademark a particular strain, we can help with claiming intellectual property. For example, if you want to trademark, register or patent a new product, it will be good to have more data. More data allows for better description of your product and the ability to prove that it is yours.
Leo: One thing that I think is a very interesting use case is proving the appellations. It is our understanding that California rolled out a procedure for growers to claim an appellation, but with strict rules around it. Within those rules, they need to prove uniqueness of growing products in specific regions. The GCxGC/MS can help in proving uniqueness by growing two different strains in two different regions, mapping out the differences and seeing what makes a region’s cannabis unique. It’s valuable for growers in California, Oregon, Colorado to be able to prove how unique their products are. To prove the differences between cannabis grown in Northern California versus plants grown along the Central Coast. And of course, for people across the world to be able to really tell a story and prove what makes their cannabis different and special. To be able to authenticate and understand, we need to have more comprehensive data about properties in those strains. It could be terpenes, it could be esters or thiols. That’s what we’re excited about.
Aaron: From your perspective, what are some of the biggest challenges and opportunities ahead for the cannabis industry?
Aldwin: Getting ready for federal legalization is both a challenge and opportunity. A challenge because when it is federally legal, there will be more regulations and more regulators. It is also a challenge because there will be more businesses, more competition, that might get into the industry. It is opening up to other players, much bigger players. Big tobacco, mega labs and massive diagnostic testing companies might participate, which will be a challenge for us.
But it’s also an opportunity for us to serve more customers, to be more established at the federal level, to move to interstate commerce. The opportunity is to be ready here and now while other people are not here yet.
Another challenge and opportunity is education. Educating consumers and non-consumers. We have to realize and accept that cannabis is not for everybody, but everyone is a stakeholder, because they are our neighbors, parents or part of the medical establishment. It would be a disservice not to educate the non-consumers.
The medical establishment, they don’t have to be consumers but they need to know about cannabis. They don’t know as much as they should about cannabis and they need to know more, like how it could affect their patients for better or for worse, so they know how to help their patients better. There could be drug interactions that could affect the potency of other drugs. They need to know these things. Educating them about cannabis is a challenge. It’s also an opportunity for us to now come in and say that cannabis is here to stay and be consumed by more and more people, so we better know how to deal with it from a medical perspective.“This bucking bronco of a growth style will throw a lot of people off. We need to figure out what we can grab on to and ride out these waves.”
Law enforcement needs to be educated too. What THC level in the blood indicates impairment? It is still a challenge because we’re not there yet, we don’t have that answer quite yet. And it’s an opportunity to help educate and to find more answers for these stakeholders, so we can have regulations that make sense.
Leo: To Aldwin’s point, the biggest opportunity comes along with federal legalization as well as expanding the customer base beyond the traditional market. Since adult use was legalized in CA, we haven’t yet seen the significant expansion of the consumer population. We’re primarily seeing a legal serving of the market that already existed before legalization.
The reality is cannabis can be used in different ways than what we think of. We know it has medical benefits and we know it is enjoyed recreationally by people looking for high THC content and the highest high. But there is also this middle ground, much like the difference between drinking moonshine and having a glass of wine at dinner. The wine at dinner industry is much bigger than the mason jar moonshine industry. That’s really where the opportunity is. What’s the appeal to the broader market? That will be a big challenge, but it’s inevitable. It comes from everything we’ve talked about today, consistency in products, educating people about cannabis, normalizing it to a certain degree, varietals and appellations.
As an entrepreneur, I’m looking at this from a business perspective. Everyone talks about the hockey stick growth chart, but it is a very wavy hockey stick. I expect to see very significant growth in the industry for a while, but it will have a lot of peaks and valleys. It’ll essentially be whiplash. We are seeing this in California right now, with sky high prices in flower last year down to bottom of the barrel prices this year. We have to all figure out how to hang on. This bucking bronco of a growth style will throw a lot of people off. We need to figure out what we can grab on to and ride out these waves. The good ones will be fun and the bad ones will be painful and we know they are coming again and again and again. That’s the biggest challenge. People say ‘expect tomorrow to look a lot like today,’ but you really can’t expect tomorrow to look anything like today in the cannabis industry. Tomorrow will be totally different from today. We need to figure out, within all this chaos, what can we hang on to and keep riding the upward trajectory without getting thrown off the bronco.